Review
Pace-of-Life and Developmental Programming of Aging
Authors
Andrzej Bartke,1,* Erin R. Hascup,2 Kevin N. Hascup,2,3 Yun Zhu,1 and Rong Yuan1
1Department of Internal Medicine, Southern Illinois University School of Medicine, Springfield, IL, USA
2Dale and Deborah Smith Center for Alzheimer’s Research and Treatment, Department of Pharmacology, Southern Illinois University School of Medicine, Springfield, IL, USA
3Department of Medical Microbiology, Immunology and Cell Biology, Southern Illinois University School of Medicine, Springfield, IL, USA
DOI:https://doi.org/10.59368/agingbio.20260044
Received: 8/20/2025, Revised: 2/20/2026, Accepted: 3/26/2026, Published: 4/21/2026
Abstract
The rate of aging, the risk of chronic disease, and the length of life can be profoundly influenced by environmental factors acting during both prenatal and postnatal development. Analysis of life histories of organisms living under natural conditions identified many examples of reciprocal relationship between pace-of-life (POL) and longevity. In mammals, the term POL is used to describe the rate of growth, the age of puberty, the early life fecundity, and the metabolic rate. The reciprocal relationship between POL and longevity is supported by studies in laboratory mice. In these animals, pharmacological suppression of postnatal growth can extend longevity. Moreover, in Prop1df mice genetically predisposed to exceptional longevity, experimental acceleration of growth during adolescence can shorten their life expectancy. Available data suggest that the mechanistic links between POL and aging include epigenetic regulation of the expression of genes impacting glucose homeostasis, energy and lipid metabolism, inflammation, and likely also other characteristics known to impact longevity. We conclude that POL during childhood and adolescence may represent a relatively accessible and practical target for life style and pharmacological interventions aimed at improving adult health and life expectancy.
Introduction
There is considerable evidence that adult phenotypic characteristics, including risk of chronic disease and other aging-related traits, can be altered by circumstances and events during early life. The widely accepted concept of Developmental Origins of Health and Disease is based primarily on studies showing that adverse conditions during pregnancy, such as famine, can increase the risk of hypertension, obesity, and cardiovascular disease in the offspring1,2. Interestingly, the magnitude and, in some cases, also the existence of this relationship depend on the postnatal environment3,4.
Numerous recent studies indicate that adult mental and physical health, as well as several measures of “biological age,” can be related not only to prenatal events but also to adversities during childhood and adolescence5–8. Association of the suppression of postnatal growth with extended longevity was seen in some of the earliest studies of the effects of calorie restriction in laboratory rodents9. Severe restriction of food intake in immature rats reduced their growth but increased their lifespan9. Similarly, male 129/Sv mice treated with the antidiabetic drug metformin during the first week of postnatal life consumed less food, weighed less, and lived longer than the control animals10.
Further evidence for the association of longevity with growth and maturation can be found in the studies of aging and maximal lifespan in organisms living in their natural habitat. These studies provide numerous examples that delayed growth and maturation are often associated with increased longevity compared to rapid growth and early maturation11,12. Differences in the pace-of-life (POL), as defined by growth, development, age of sexual maturation, fecundity, and metabolic rate, are generally inversely related to differences in longevity, with faster POL predicting shorter life11–13.
Interestingly, the negative association of longevity with POL, defined primarily by growth rate, also applies to forest trees13. A causal relationship between at least one of the defining characteristics of POL and aging was demonstrated by showing that selection of fruit flies for late reproduction produced populations with extended longevity14.
In this brief review, we will focus on the recent evidence that aging and longevity can be influenced (“programmed”) by conditions and interventions during early postnatal life and on their apparent mechanisms. Our emphasis on the period of postnatal life corresponding to childhood and adolescence stems from the belief that lifestyle and, perhaps, also pharmacological interventions aimed at slowing the rate of aging may be ethically acceptable and practical during this period.
Socioeconomical Status and Developmental Programming of Aging
There is considerable evidence that adult health-related and aging-related traits can be modified by adversities during childhood and adolescence5–8. Risk of adverse conditions during development is related to socioeconomical status. Although specific mechanisms linking socioeconomical status and health have not been clearly identified, they almost certainly include lifestyle factors such as quality of the diet, opportunities for exercise, smoking and secondary smoke exposure, as well as awareness of the importance of these factors and access to healthcare. Studies of these relationships revealed that various measures of social position (income, education, postal code) are associated with health and longevity5,7. Against this background, a recent study of Marston and colleagues15 addressed the association of family affluence with leukocyte telomere length, a putative early marker of biological aging, and with the production of cortisol, a key hormonal index of chronic stress. This study involved over one thousand children, aged between 5 and 12 y, from six European countries. Cortisol production was calculated from the levels of this hormone and its multiple metabolites in morning and evening urine samples. Higher affluence was associated with longer telomeres and lower cortisol production. However, these two parameters were not significantly associated, and analysis of the data did not identify cortisol as a mediator of the effects of affluence on telomere length. In as much as telomere length can be considered a measure or a proxy index of biological age, these results indicate that social advantage of greater family affluence can set the trajectory of aging to a slower pace starting in childhood. This is a very important observation because accelerated and/or earlier aging could represent a key mechanism of the negative impact of social disadvantage and early-life adversities on adult health. Age is an important and, in many cases, dominant risk factor for most chronic metabolic and degenerative diseases, as well as susceptibility to infectious agents. The mechanisms underpinning relationships between early-life environmental stressors and aging remain to be clearly identified. Results of the study by Marston et al. did not support the possible role of the chronic stress-related alterations in cortisol production in mediating the effects of family affluence on the examined measure of biological aging. Further studies will be needed to relate these findings to the reported effects of social stressors on telomere length16 and to identify mechanisms involved.
POL, Energy Metabolism, and Longevity
In ecological studies, species with a fast POL are generally short-lived, while those with a slow POL are long-lived. Since POL is defined in terms of growth, development, onset of reproduction, lifetime fecundity, and metabolic rate, long-lived animals would be expected to have low metabolic rates. This relationship is exemplified in the generally positive relationship between adult size and longevity in comparisons between different species and the classical “mouse-elephant curve,” in which large, long-lived animals have a lower basal metabolic rate than the small, short-lived species. Although differences in metabolic rate appear to offer an intuitive explanation for the many examples of the association of larger body size with longer life, the relationship of basal metabolic rate to longevity was not evident in data corrected for body mass and phylogeny12. Moreover, there are many exceptions, particularly in comparisons of longevity within, rather than between, species. For example, studies of energy metabolism in animals, which are long-lived because of genetic suppression of somatotropic signaling, demonstrated increased, rather than reduced, total energy expenditure as measured by daily oxygen consumption (or heat production) per gram of body weight17,18. One of the possible reasons for the discrepancy between these sets of data is the difficulty of relating basal metabolic rate, which is typically measured at rest or during sleep and at thermoneutral temperature, with the average metabolic rate of freely moving animals housed at a standard room temperature, which in the case of mice is considerably below thermoneutrality. Under these conditions, diminutive, growth hormone (GH)-deficient and GH-resistant mice lose much body heat by radiation, and a large portion of their metabolic rate represents energy devoted to thermogenesis needed to maintain normal body temperature. It should also be pointed out that the total daily oxygen consumption, that is, oxygen consumption expressed per animal rather than per gram body weight, is reduced in long-lived GH-related mutants19. Therefore, we believe that in spite of the increased energy expenditure per unit of body mass, it is justified to consider these mutants as having a slow POL and providing yet another example of the association of slow POL with extended longevity. Housing diminutive GHRKO mice at thermoneutral temperature (30°C) reduced VO2 during the active phase. This was associated with extension of longevity in females20.
The negative association of metabolic rate, one of the defining characteristics of POL, with longevity is further supported by recent studies in primates. Primates exhibit many characteristics of a slow POL, live longer than would be predicted based on their body mass, and have lower total energy expenditure than expected for their size21. In humans, in which the discrepancy between the maximal longevity and longevity predicted on the basis of body weight is particularly pronounced, slow growth (a key feature of slow POL) may be due in part to the energy needed to support brain development and function22. In further support of a negative association of metabolic rate and longevity, hypermetabolism due to OxPhos defects in cultured cells or mitochondrial disease in patients was recently reported to be associated with reduced lifespans23. Perhaps mitochondrial inefficiency and stress associated with dysfunctional energy metabolism contribute to accelerated aging. More broadly, hyperfunction was suggested to promote aging and to underlie the negative association of mTOR Complex 1 signaling and longevity, which is seen across the tree of life24,25.
Mechanistic Links between Development and Aging
Evidence for the negative association of the rate of development and longevity brings up a number of questions about the specific elements of this association and the mechanisms involved. One could ask whether the rate of aging and the lifespan are primarily related to the age of sexual maturation. This could involve the extensively researched and much-debated trade-offs between reproduction and aging. Alternately, aging and longevity could be driven by the rate of somatic growth, the timing of specific growth stages, or the maturation of a particular organ system, most likely the brain. In addition, differences in the rate of aging and in longevity could be related to the final outcome of growth and maturation processes, namely the adult body size, or the relative or absolute size of the brain. The possible mechanisms that could link brain size with aging are unclear and presumably mostly indirect, such as the impact of cognitive processes, communication, and social organization on life expectancy in primates versus other mammals. The relationship of adult body size to aging is suggested by countless datasets but is unlikely to be causal since it is generally opposite in comparisons among the members of the same species versus comparisons of different species within larger taxonomic groups such as mammals or birds. Separating the role of growth from the role of maturation in the control of aging is difficult because these processes occur concurrently, and sexual maturation is generally reached after attainment of a specific body weight and/or composition. This complex issue was addressed in a recent study of Kang and colleagues26 using mutants of a fruit fly, Drosophila melanogaster. In this insect, developmental timing is controlled primarily by the prothoracicotropic hormone, PTTH. This hormone does not affect growth rate. Loss-of-function mutants of the PTTH gene are characterized by developmental delay, no changes in the rate of growth, an increase in adult body size, and significantly extended longevity26. These effects were shown to involve the action of a key insect hormone, ecdysone, and suppression of inflammatory (NF-kB-homologous) signaling during larval stages of development26. Thus, these mutants uncouple developmental time from growth rate and provide evidence for a mechanistic link of the former, but not the latter, to adult life span. Studies in mice treated early in life with a putative antiaging drug, metformin, suggest that the timing of female sexual maturation can be uncoupled from somatic growth also in mammals27, but the impact of this dissociation on aging and longevity remains to be explored.
In several studies, the impact of early (preweaning) nutrition on adult health and aging was investigated by altering the number of suckling pups. In the Miller laboratory, pups were added to litters of genetically heterogeneous UM-HET3 mice to reduce nutrient availability during suckling28. Pups from these “crowded litters” weighed less at weaning than pups from smaller (control) litters, and this difference persisted into adulthood28. Moreover, they were characterized by reduced hypothalamic astrogliosis and by increased mean and maximal lifespan29 in keeping with the negative association of POL and aging.
In studies directed at developing an animal model of childhood obesity, the litter size of Wistar rats was reduced to 2–4 pups per dam30. The resulting overfeeding led to increased visceral adiposity, insulin resistance, dysregulation of glycemic homeostasis, dyslipidemia, liver steatosis, and cardiovascular dysfunction in adulthood30. These alterations could be prevented by treatment with fenofibrate during lactation, implicating a role of reduced PPAR alpha activity in the developmental programming of the examined adult metabolic characteristics30. The effects of overfeeding during early life on longevity have not been reported in these studies, but metabolic characteristics of the overfed animals are consistent with the prediction of accelerated aging and reduced life span. Thus, data from these studies provide yet another example of the detrimental impact of accelerating POL on adult health and aging.
Identifying causative links among POL, healthy aging, and longevity in nutritional studies and in epidemiological data is complicated by the overlap and interplay of factors that affect growth and sexual maturation as well as the risk of obesity. These factors include various elements of life style, primarily food intake and energy expenditure. Obesity can affect both healthspan and lifespan by its well-documented impact on the risk of metabolic syndrome, type 2 diabetes, and other chronic diseases31. Increased risk of diabetes is particularly relevant in this context since this disease mimics many symptoms and diseases of aging, including increased incidence of frailty, cardiovascular disease, cancer, and Alzheimer’s disease32,33. Childhood obesity is associated with advanced sexual maturation, a key element of fast POL34.
A recent study by Kolb and his colleagues demonstrated that selective inhibition of early postnatal growth can have a major impact on adult phenotype and longevity35. This study utilized mice with inactivation of the alpha-casein gene and consequent changes in the amount and composition of milk proteins. Pups nursed by alpha-casein-deficient dams grew very poorly, and at weaning were much smaller than pups nursed by dams fed a low protein diet and from pups in experimentally enlarged (“crowded”) litters. The difference in body weight between pups nursed by casein-deficient versus wild-type females became less pronounced when the animals started to eat solid food but did not disappear. Interestingly, marked reduction of the expression of IGF-1 and GH at 21 days of age and concomitant increase in the expression of IGFBP2 were no longer evident in animals that reached the age of 100 days. The authors emphasized the contrast between these findings and the lifelong deficiency of somatotropic signaling in Prop1df (Ames dwarf) mice and other GH-related, long-lived mutants. Longevity was significantly extended in pups nursed by alpha-casein-deficient dams, and it was shown by cross-fostering that this effect was due entirely to the genotype of the dam with no impact of the genotype of the pups. Extension of longevity was seen in both sexes, but it was greater in females (23%) than in males (16%). Studies of body composition revealed an unexpected decrease in adiposity and in the size of the adipocytes. There was little change in brain growth, resulting in an increase in relative brain weight. Comparison with the findings in animals exposed to calorie restriction suggests that extended longevity could be traced to reductions in adult body weight and in adiposity. Analysis of hepatic gene expression identified other potential mechanisms of extended longevity, including an increase in the markers of xenobiotic metabolism, suppression of mTOR Complex 1 signaling, and activation of Nrf/SKN-1, GCN2-ATF4, and FGF 2135. The effects of maternal alpha-casein deficiency on the longevity of the offspring suggest that growth rate during lactation is a very important, likely a key, element linking POL to aging and lifespan.
Lean phenotypes of mice experiencing transient, early-life suppression of somatotropic signaling as a result of maternal alpha-casein deficiency contrast with the effects of lifelong GH deficiency or resistance on body composition. Hypopituitary, GH-deficient mice and mice with deletion of GH receptor (GHR) have markedly increased amounts of white adipose tissue (WAT), particularly in the subcutaneous depots such as inguinal WAT36,37. Increased adiposity is also seen in patients with isolated GH deficiency and in GH-resistant patients with Laron syndrome38–40. In both human subjects and mice with these hereditary conditions, increased adiposity is believed to be due to the loss of the well-documented lipolytic action of GH. Surprisingly, reducing GH signaling only during early postnatal life by maternal casein deficiency does not duplicate these changes in body composition but instead leads to leanness during adult life. The mechanisms responsible for reduced WAT deposition in these animals remain to be elucidated. Further work will also be needed to explain why longevity is extended in both lean progeny of casein-deficient mice and in corpulent GH-resistant and GH-deficient mutants. The unexpected coexistence of increased adiposity with extended healthspan and lifespan in these animals has been related to the altered anti- rather than pro-inflammatory profile of cytokine secretion by their WAT41,42. Reduced levels of IL-6 and TNF alpha, together with increased levels of adiponectin, likely contribute to improved metabolic health and increased life span of GH-deficient and GH-resistant mice.
In a recent study, Shindyapina and her colleagues examined the effects of reducing postnatal growth rate on aging and longevity by treating lactating UM-HET3 mice and their weanlings with rapamycin, an inhibitor of mTOR Compex1 signaling43. Exposure to rapamycin during the first 45 days of life caused the expected reduction in growth rate, and the animals remained smaller than controls after the treatment was stopped. In these rapamycin-treated mice, the reproductive age was delayed while measurements of glucose and insulin tolerance, gait speed, and various symptoms of frailty indicated preservation of health. The median lifespan was increased by approximately 10%. This primarily reflected the extension of longevity in males, while a similar trend in females was small and not statistically significant43. Results of the study by Shandyapina et al.43 provide important evidence that experimentally induced slowing of early-life growth and development can extend longevity in normal, genetically heterogeneous mammals.
Results of studies in a different strain of mice, which were conducted in another laboratory and reported in the same year44, provided compelling evidence for the unique importance of early-life events in the determination of longevity. In this work, outbred CD1 mice were given daily rapamycin injections between 4 and 30 or between 30 and 60 days of age. Treatment started on day four significantly increased lifespan in both females and males, while treatment started on day 30 did not. The early-life rapamycin treatment had a greater impact on the adult body weight than treatment started later in life and induced changes in the profile of hepatic gene expression, which were different from the alterations detected in mice treated between 30 and 60 days of age. Since many of the gene expression changes induced by the early rapamycin treatment did not persist after the treatment was stopped, it was proposed that some sort of a “memory” process impacts the trajectory of aging. The authors suggested that upregulation of the expression of sulfotransferases by early-life rapamycin treatment may represent one of the mechanisms of the extension of longevity. In support of this argument, they detected upregulation of sulfotransferase expression and extension of longevity also in Drosophila treated with rapamycin during larval development. Moreover, transient larval overexpression of dST1, a gene that was acutely upregulated by rapamycin in larvae but not in adults, was effective in increasing Drosophila lifespan44.
While a number of studies sought to identify mechanisms linking POL to the rate of aging and the regulation of longevity, there is little information on the question of how POL and the corresponding reproductive strategies are evolutionarily shaped by the environment. This important question was addressed by Ahi and colleagues in a recent review article45. These investigators proposed that the Hippo signaling pathway links environmental factors such as ambient temperature and food availability to regulation of growth, sexual maturation, and reproduction via metabolic sensors (including AMPK and mTOR) and hormones known to affect growth, timing of puberty, and reproduction. Together with emerging evidence for the involvement of epigenetic mechanisms46, studies of this signaling pathway will likely lead to a better understanding of the role of POL in the adaptation to environmental constraints and the functioning of various trade-offs and feedback loops that connect POL with the time course of aging.
GH Signaling, POL, and Longevity
The role of POL in the control of aging and longevity is strongly supported by studies in mice with mutations blocking various steps of the biosynthesis, secretion, or actions of pituitary GH, a key regulator of somatic growth and reproductive development. These mice are characterized by prominent features of slow POL: slow growth, delayed sexual maturation, reduced fecundity, and extended longevity40,47,48. Importantly, there is evidence that in these mice the links between reduced GH signaling and extended longevity, as well as adult characteristics related to metabolic health, are causal rather than merely correlative. In GH-deficient Prop1df (Ames dwarf) mice, GH replacement therapy can reverse (i.e., normalize or “rescue”) many of these characteristics and shorten life49–51 (
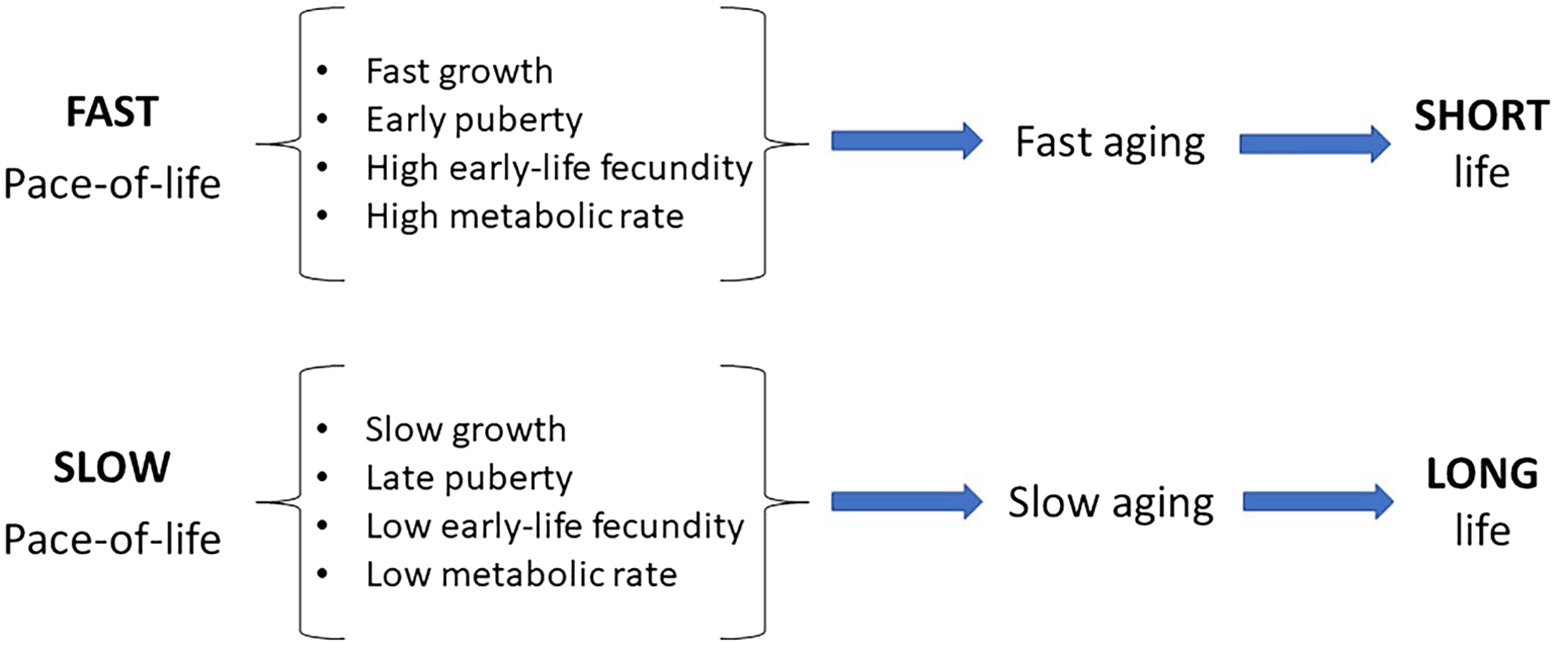
Differences in the pace-of-life are associated with differences in the rate of aging and longevity.
Additional important, although indirect, evidence for the importance of early-life GH signaling on aging and longevity comes from comparison of the effects of congenital versus induced (adult) GHR deficiency in laboratory mice. Studies in the Kopchick laboratory provided evidence that while congenital GHR deficiency produces impressive extension of longevity in both sexes, disruption of GHR at the age of six weeks or six months produces a smaller increase in the lifespan, which is significant only in females56,57. A very interesting extension of these studies was recently reported by Poudel et al.58. In this work, GHR deficiency was induced at 12 months of age, and the animals were examined one year later. GHR-deficient (iGHRKO 12-24) mice had reduced hypothalamic inflammation, no changes in markers of systemic inflammation, impaired bone morphology, and feminization of hepatic transcriptome in males. The authors interpreted these findings as the induction of a mixture of pro- and antiaging effects and concluded that “inhibiting the GH/IGF-1 axis during aging only partially preserves the beneficial healthspan effects observed with congenital GH deficiency58.”
Conclusions
What we are proposing is a major role of developmental programming in shaping the trajectory of aging. More specifically, we are arguing that a combination of developmental and metabolic traits, collectively known as pace-of-life (POL), predicts age-related physiological changes, risk of chronic disease, and longevity. We feel that this proposal does not contradict the generally accepted view that aging is not programmed when the term “programming” is used to denote the existence of specific mechanisms that have been evolutionally developed to drive aging.
The links among POL, aging, and longevity are almost certainly epigenetic, likely mediated by DNA methylation, histone acetylation, and consequent alterations in expression of multiple genes. We acknowledge that in addition to POL, aging is accompanied and/or driven by an almost endless list of environmental influences, as well as cellular and organismal changes (as exemplified by the hallmarks or pillars of aging). POL emerges as one of the quantitatively important determinants of the time course of aging as well as longevity. Importantly, this conclusion is based on data derived from studies of environmental impacts (including parental, social, and nutritional influences), as well as experimentally induced genetic alterations and comparisons of the life history of different species living in their natural environment.
In terms of practical recommendations for human nutrition, the relationship of adult health and longevity to POL is difficult to dissociate from the well-documented role of caloric intake and food quality in the epidemics of obesity currently affecting most human populations. Findings presented in this brief review suggest that avoiding excessive food intake and maintaining a healthy composition of the diet during the period of rapid growth before and around the time of puberty may not only help in maintaining a healthy body weight but also prevent early aging and the associated increase in the risk of chronic disease.
Acknowledgments
We apologize to those whose work pertinent to the issues discussed was not cited due to limitations of the format or to inadvertent omissions. We are grateful for editorial assistance provided by Lisa Hensley. This work was supported by Hevolution Foundation HF-GRO-23-1199066-37, the William E. McElroy Charitable Foundation, and the Geriatrics Research Initiative at Southern Illinois University School of Medicine [AB], and the William E. McElroy Charitable Foundation [RY].
Conflict of Interest
The authors declare that they have no actual or apparent conflict of interest between authorship of this study and any other activities.