Research Paper
Translational Changes upon Aging and Dietary Restriction in Progeroid DNA-Repair-Deficient Mice
Authors
Ivar van Galen,1,2 Rutger A. Ozinga,1,2 Damon A. Hofman,1,2 Jip van Dinter,1,2 Sem A. G. Engels,1 Kimberly Smit,1,2 Sebastiaan van Heesch,1,2 Jan H. J. Hoeijmakers,1,2,3,4 and Wilbert P. Vermeij,1,2,*
1Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands
2Oncode Institute, Utrecht, The Netherlands
3Department of Molecular Genetics, Erasmus MC Cancer Institute, Erasmus University Medical Center Rotterdam, Rotterdam, The Netherlands
4Institute for Genome Stability in Ageing and Disease, Cologne Excellence Cluster for Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Cologne, Germany
*Corresponding author: W.P.Vermeij@prinsesmaximacentrum.nl
DOI:https://doi.org/10.59368/agingbio.20260043
Received: 9/5/2025, Revised: 12/19/2025, Accepted: 12/28/2025, Published: 2/3/2026
Full Article | PDF | Supplementary
Abstract
Aging is a complex multifactorial phenomenon largely driven by damaged macromolecules. We showed recently that with aging, time- and exposure-dependent accumulation of DNA damage derails the basal process of transcription, physically stalling RNA polymerase, lowering and skewing the transcriptional landscape in a gene-length-dependent fashion. However, how this influences the translational output and whether translation is similarly affected is largely unknown. Here, we present a parallel analysis of transcriptional and translational liver profiles from the well-characterized Ercc1Δ/− progeroid, DNA repair-deficient mouse model compared to wild-type under ad libitum conditions and upon dietary restriction (DR), which strongly delays aging in this mutant.
Using ribosome profiling, we found that transcriptional changes during accelerated, normal, and delayed aging are largely preserved at the translational level, ruling out a major translational impact on gene expression in aging. Moreover, in both Ercc1Δ/− and aged wild-type mice there was a prioritization of inflammation, metabolic redesign, and expression of translation initiation factors, along with a shift in codon occupancy. While translation initiation factors were further increased by DR, absolute codon occupancy was partially normalized, showing a discordant response. In addition, increased ribosomal pausing and a relative reduction of upstream open reading frame expression were both further intensified by DR.
Together these data infer a fine-tuning of the translational output, for example, by regulating upstream open reading frames under various cellular stress situations. This study uncovers a complex interplay between DR, DNA damage, aging, and translational regulation, highlighting the potential of DR to modify DNA damage-driven translational dynamics during aging.
Introduction
Genomic instability has been posited as a central hallmark of aging and is characterized by the accumulation of a wide range of DNA lesions1,2. While most DNA lesions are efficiently resolved by their respective repair systems, some damages escape recognition, are irreparable or repaired incorrectly, leading to the accumulation of mutations and persistent DNA lesions with age1,3. This DNA damage drives systemic aging by physically blocking transcription, leading to a lower and particularly imbalanced gene-length-dependent transcriptional output3–5, and since transcription is essential for every cellular process, this age-related phenomenon of genome-wide transcription stress indirectly affects major aging hallmarks4,6,7.
Next to a disbalanced transcriptional output, aging does induce a general decoupling between the transcriptome and proteome8–11. Furthermore, aging is associated with a dysregulation of proteo-stasis and a reduction in protein biosynthesis across diverse tissues and organisms12,13 and can be further altered during age-related diseases14,15. Age-driven changes also extend to the stoichiometry of protein complexes, such as the cytosolic ribosome complex, where both a dysregulation between transcript and protein levels and a stoichiometric loss between subunits have been reported10. In addition, aging influences translational dynamics, leading to increased ribosome pausing and collisions, shifts in transcript coverage at start and stop codons, and alterations in the translation of upstream open reading frames (uORFs)16–19, culminating in a reduced and altered translational profile20. Interestingly, DNA damage has been implicated in many of the above processes, including derailed transcription4, inhibited translation21, and ribosome stalling22, subsequently contributing to inflammation23. However, how DNA damage impacts translation in relation to the before mentioned age-related gene-length-dependent transcriptional imbalance is largely unknown.
To assess the effect of aging and accumulating DNA damage at the level of translation, we compared side-by-side mRNA- and Ribo-seq. For this, we used the well-characterized Ercc1Δ/− DNA-repair-deficient progeroid mouse model5,24–26. These mice harbor a single allele coding for a truncated yet partially active ERCC1 variant, a crucial factor involved in multiple DNA repair pathways. The dysfunctional ERCC1 in these mutants leads to a broad variety of unrepaired persisting genomic lesions, accelerating the aging process systemically and limiting their lifespan to 4–6 months27. Remarkably, a 30% dietary restriction (DR) regimen, the only evolutionary conserved intervention for extending health and lifespan across multiple species, has been shown to partially mitigate transcriptional aberrations in Ercc1Δ/− mice, prolonging their lifespan up to 200% and strongly delaying the onset of numerous age-related phenotypes5.
To elucidate if and how transcriptional changes in progeroid Ercc1Δ/− mice are altered at the translational level, we utilized ribosome profiling, a technique that captures a snapshot of active translation. In addition, we investigated how DR, which partially alleviates DNA damage and consequently decelerates aging in these animals, modulates these dynamics, thereby providing insights into the interplay between diet, DNA damage, and translation in the context of DNA damage-driven aging.
Results
Transcription and translation are influenced by genotype and diet
To investigate active translation during DNA-damage-driven accelerated aging and the anti-aging effects of dietary restriction (DR), we selected liver as the primary organ of interest due to its key role in metabolism, detoxification, and the extensively characterized transcriptional landscape during aging in both wild-type (WT) and Ercc1Δ/− progeroid DNA-repair-deficient mice4,5,28,29. To capture the progeroid aging phenotype effectively, we focused our analysis on 16-week-old male mice, an age at which Ercc1Δ/− mice exhibit pronounced aging characteristics but prior to becoming overtly moribund27 and the benefits of DR and weight changes are very prominent5. The experimental setup encompassed: ad libitum (AL) feeding and 30% DR, maintained over the last ∼8 weeks (
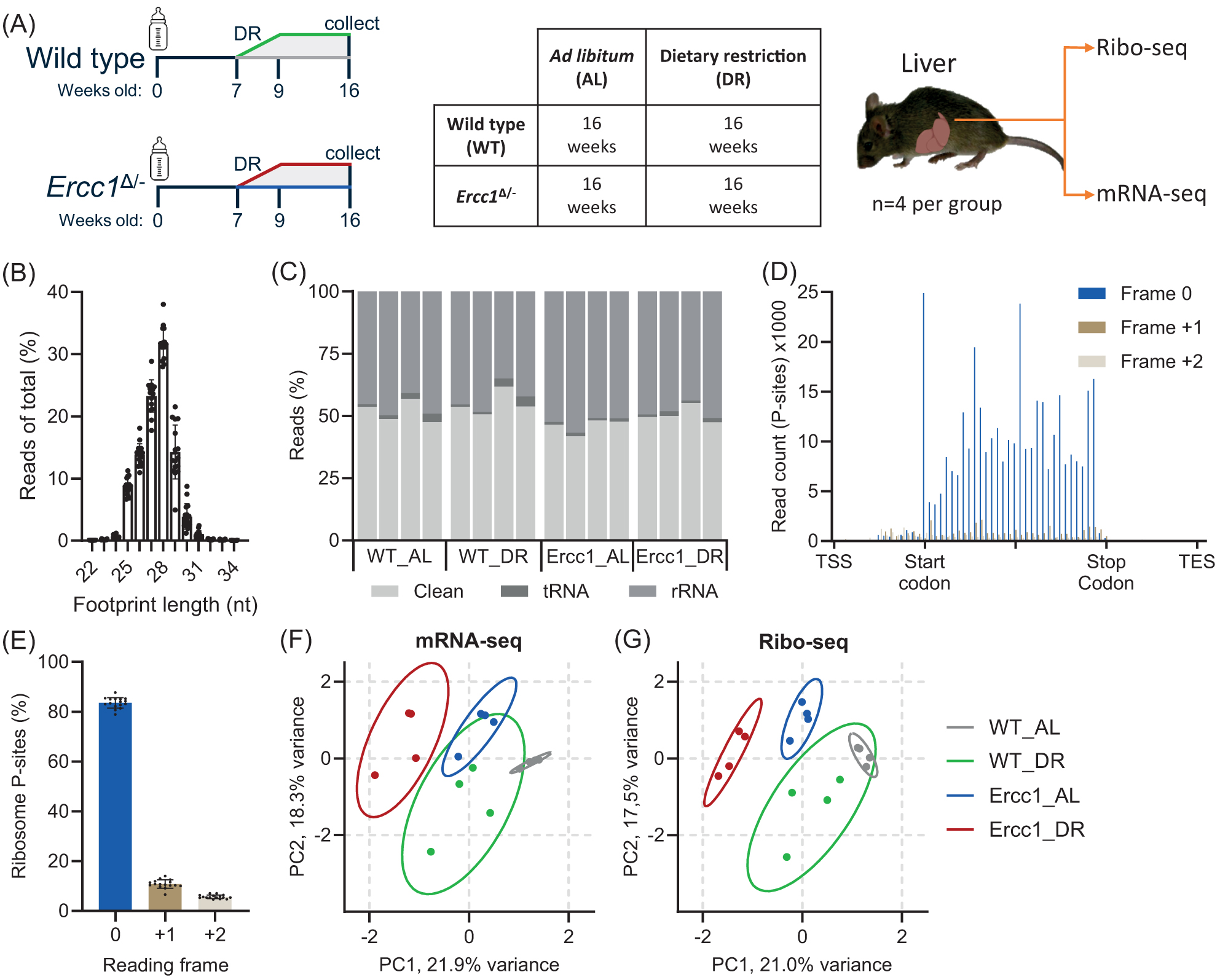
(A) Schematic representation of experimental setup and tissue acquisition from male wild-type and Ercc1Δ/− mice under AL and DR conditions: WT_AL (gray); WT_DR (green); Ercc1_AL (blue); and Ercc1_DR (red). Food was provided to the animals just before the start of the dark (active) period, Zeitgeber Time (ZT) 12:00, except for the day of sacrifice, when no food was given prior to collect between ZT13 and ZT16. (B) Footprint length distribution compared to total reads. (C) Quantification of RNA contaminants. Reads that were not tRNA or rRNA were considered “clean.” (D) Gene meta-analysis of p-site-specific frame preference between Transcription Start Site (TSS) and Transcription End Site (TES). (E) Quantification of p-site frame preference indicative of footprint periodicity. (F,G) PCA plots with 95% confidence intervals for mRNA- (F) and Ribo-seq (G). Values are mean ± SD.
Our ribosome profiling data showed a clear 28 nt footprint preference, with about half of all reads considered “clean” as they did not map to tRNA or rRNA sequences (
Principal Component Analysis (PCA) of both mRNA and ribo-seq data showed distinct clustering and uniformity within and between groups, with primary differentiation based on genotype and diet (
ERCC1-deficiency modifies metabolic and inflammatory pathways
When directionality and fold-change differences between Ercc1Δ/−_AL and WT_AL at the transcriptional level were compared with those at the translational level, a high degree of concordance was observed (
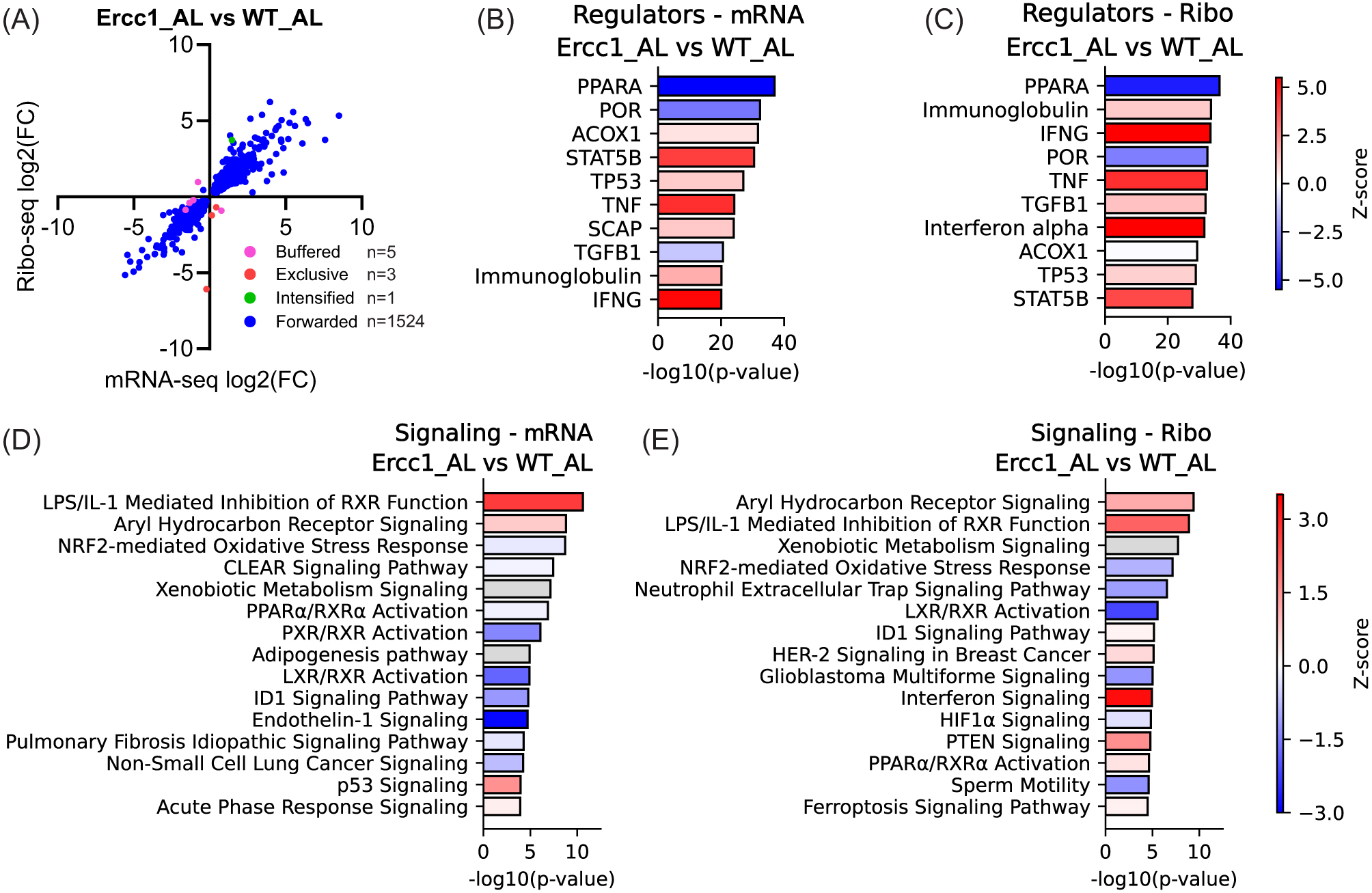
(A) Scatter plot of gene expression (n = 1533) in Ercc1Δ/− mice, comparing transcriptional (mRNA-seq) and translational (Ribo-seq) levels. Forwarded genes have a significant change in mRNA and ribosome-protected fragments (RPF) at the same rate, with no significant change in translational efficiency (TE). Exclusive genes are regulated only at the translational level, intensified genes have a significant change in TE that acts with the effect of transcription, and buffered genes have a significant change in TE that counteracts the change in RNA. (B–E) Upstream regulator (B,C) and signaling pathway (D,E) analysis based on differentially expressed genes in AL-fed Ercc1Δ/− mice compared to WT at the mRNA (B,D) and Ribo-seq (C,E) level. Numerical parameters of pathways and upstream regulators were calculated with Ingenuity Pathway Analysis (IPA), and subsidiary pathways were collapsed if top parent pathways were present. Red means upregulated, white indicates pathways with z-scores near zero or those ineligible due to a limited number of hits, blue means downregulated, and gray means that no activity prediction could be made for this pathway.
ERCC1-deficiency alters translational dynamics and open reading frame translation
Natural aging affects various aspects of translation, including fidelity and kinetics18. Evaluating the expression of essential components of the translation machinery in Ercc1Δ/−_AL mice revealed a significant increase in translation initiation factors (
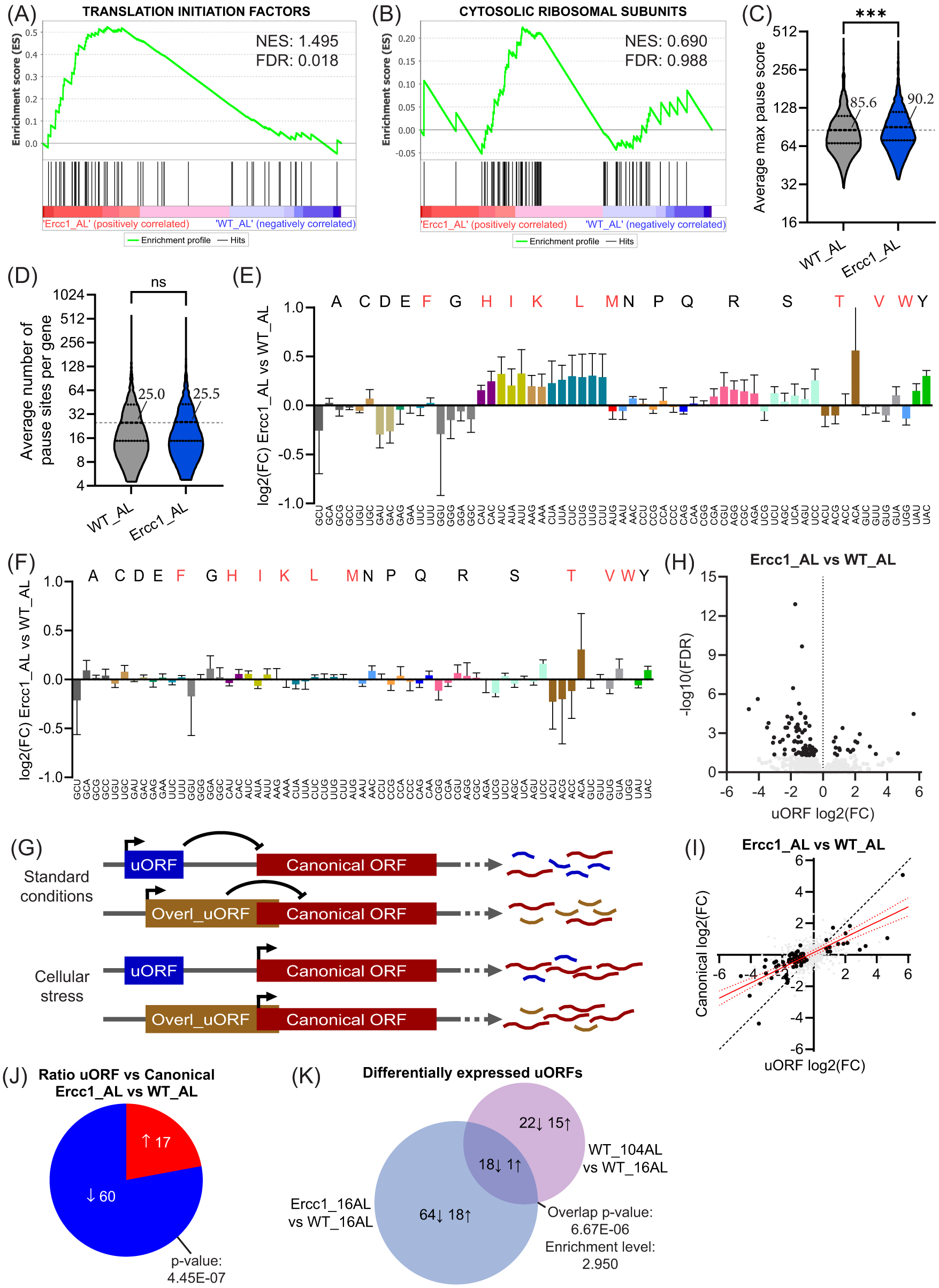
(A,B) GSEA enrichment graphs of translation initiation factors(A) and cytosolic ribosomal subunits (B), based on ribosome profiling data (C,D) Violin plots depicting the max pause score intensity (C) and the mean number of pause sites found per gene transcript averaged per group (D). Highlighted intensity values represent median intensity. (E,F) Bar graph plot showing absolute (E) and normalized (F) P-site codon occupancy log2 fold changes in Ercc1Δ/− as compared to WT with essential amino acids being indicated in red and error bars depicting SD. (G) Simplified graphical representation of the regulatory function of (overlapping) upstream open reading frames (uORFs) on a transcript’s canonical ORF. (H) Vulcano plot of (overlapping) uORF expression in Ercc1Δ/−_AL as compared to WT_AL, with differentially regulated uORFs being indicated in black. (I) Scatterplot illustrating the relation between uORF and canonical fold change differences in Ercc1Δ/−_AL versus WT_AL with significantly altered uORFs being depicted in black. A regression line with a 95% confidence interval, based on the significant uORFs, is represented in red. (J) Pie chart depicting the number of uORFs that are more strongly up- or down-regulated than the canonical ORF they belong to in Ercc1Δ/−_AL as compared to WT_AL. (K) Venn diagram of the overlapping differentially regulated uORFs in Ercc1Δ/−_AL and 104-week-old WT_AL compared to young 16-week-old WT_AL mice. Arrows indicate activation or inhibition of uORF expression. Significance levels are denoted “ns” for p > 0.05 and *** for p ≤ 0.001, with comparisons made to indicated control groups.
Next, we examined ribosome pausing, an important aspect of translation kinetics. This process is characterized by a halt or significant slowdown in ribosomal movement along the mRNA strand during protein synthesis and is quantified by the distance and distribution of ribosomal footprints along the mRNA. Analysis of footprint density yielded ribosomal pause site intensity scores and the number of pause sites per gene transcript. Our data showed a significant increase in pause score intensity (
To gain further insight into translational dynamics, we assessed codon occupancy, a proxy for the frequency and duration of ribosome binding and translation of specific codons within mRNA sequences35. Absolute codon occupancy (pre-normalization for amino acid translational abundance and thus indicative of absolute changes in codon usage between groups) revealed distinct alterations in Ercc1Δ/−_AL mice as compared to their wild-type counterparts. Specifically, codons encoding many essential amino acids showed increased occupancy, in addition to nearly all codons for Arginine (R) and Serine (S) (
Next, we examined the translated rates of non-canonical and canonical open reading frames (ORFs) in Ercc1Δ/−_AL mice, as these are known to be affected by cellular stressors36,37. Upstream ORFs (uORFs), a non-canonical type of reading frame located in the 5’ untranslated region (UTR) of mRNAs, have the potential to regulate the translation of downstream canonical coding sequences (CDS). Overlapping uORFs and conventional uORFs tend to inhibit translation of the CDS (
In Ercc1Δ/−_AL, we identified 101 differentially translated uORF containing transcripts, which were generally downregulated compared to WT_AL (
Differences between Ercc1Δ/− and old WT mice
Ercc1Δ/− mice display an accelerated aging phenotype primarily driven by DNA damage, while natural aging is affected by various stressors, including DNA damage. Comparing directionality and fold-change differences of DEGs at the transcriptional and translational level between 16-week-old Ercc1Δ/− and 104-week-old WT mice showed a high degree of concordance (Fig. S4A).
In contrast, IPA upstream regulator analysis showed some disparity between the top mRNA and Ribo results (Fig. S4B,C). While the upstream regulators POR and PPARA were both predicted to be decreased in Ercc1Δ/− and old WT when compared to young WT, which has been reported before for natural aging42. Direct comparison of Ercc1Δ/− to old WT showed that POR was relatively more inhibited in Ercc1Δ/−, while PPARA was increased, potentially due to altered lipid metabolism in Ercc1Δ/− mice43. Ercc1Δ/− also showed a markedly lower activity of TGFB1 than old WT, which was more pronounced at the transcriptional level. Notably, NFE2L2 and immunoglobulin complex were predicted to be activated in Ercc1Δ/− mice when compared to old WT, in line with the elevated oxidative stress and altered inflammatory signaling reported for this model (Fig. S4B,C)44.
Analysis of metabolic processes showed that most were relatively increased in Ercc1Δ/− compared to old WT and that they centered around lipid metabolism (as indicated by cholesterol synthesis and fatty acid B-oxidation) and oxidative stress (e.g., increased glutathione-mediated detoxification; Fig. S4D,E). In addition, signaling pathways were largely lowered and characterized by LPS/IL-1 mediated inhibition, sirtuin signaling, NRF2 oxidative responses (NFE2L2), and NAFLD signaling, all strongly related to stress mechanisms and altered lipid metabolism (Fig. S4F,G)45,46. As mentioned above, there were differences in absolute codon occupancy in Ercc1Δ/− and old WT that were relatively similar when compared to young WT animals. Direct comparison of Ercc1Δ/− to old WT similarly showed that differences were limited, but that some codon groups (e.g., histidine (H), isoleucine (I), and Tyrosine (Y)) were more highly expressed in Ercc1Δ/− (Fig. S4H). Differences in relative codon occupancy were largely negligible, except for AUA, which showed a subtle increase, and AUU and UCG, which decreased in Ercc1Δ/− (Fig. S4I). Finally, uORF expression was reduced in Ercc1Δ/− mice, with a more pronounced decrease than observed for canonical ORFs (Fig. S4J,K).
DR partially reverses processes affected in Ercc1Δ/− mice
DR alleviates genotoxic stress in Ercc1Δ/− mice, likely by triggering a protective, anti-aging “survival” response boosting maintenance and resilience (e.g., antioxidant defense)5,47,48. To evaluate this scenario, we analyzed the expression and translational parameters affected in Ercc1Δ/−_DR mice and assessed DNA damage-related changes relative to AL-fed animals. Comparing DR-regulated changes in ribosome profiling data with mRNA again revealed high similarity and equal variance (
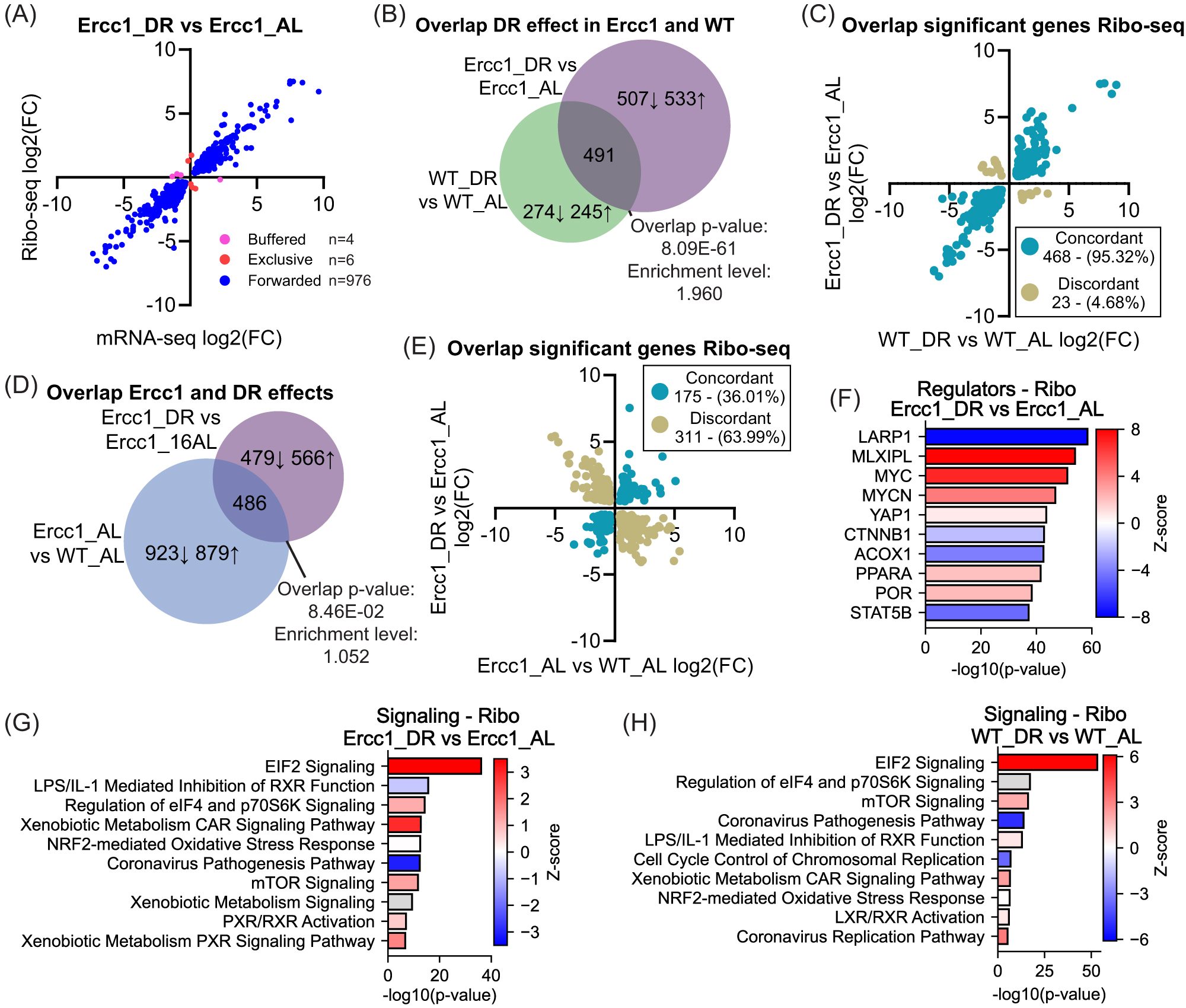
(A) Scatter plot of gene expression (n = 986) in DR-fed versus AL-fed Ercc1Δ/− mice, comparing transcriptional (mRNA-seq) and translational (Ribo-seq) levels. Forwarded genes have a significant change in mRNA and RPF at the same rate, with no significant change in translational efficiency (TE). Exclusive genes are regulated only at the translational level, and buffered genes have a significant change in TE that counteracts the change in RNA. (B,C) Venn diagram of the DEGs and their overlap due to DR in Ercc1Δ/− and WT mice as compared to AL-fed controls (B). Scatter plot of the overlapping DEGs divided between concordant and discordant expression (C). (D,E) Venn diagram of the DEGs caused by the Ercc1Δ/− mutation, the effect of DR in Ercc1Δ/−, and their overlap (D). Scatter plot of the overlapping DEGs divided between concordant and discordant expression (E). (F) Upstream regulator analysis of ribosome profiling based on differentially expressed genes in DR versus AL-fed Ercc1Δ/− mice level. (G,H) Signaling pathways altered due to DR in Ercc1Δ/− (G) and WT (H) genotypical background. Numerical parameters of pathways and upstream regulators were calculated with Ingenuity Pathway Analysis (IPA). Red means upregulated, white indicates pathways with z-scores near zero or those ineligible due to a limited number of hits, blue means downregulated, and gray means that an activity prediction could not be calculated for this pathway.
The DR-induced differentially expressed genes (DEGs) in Ercc1Δ/− and WT mice showed significant overlap (
Upstream regulator analysis of genes differentially translated in Ercc1Δ/−_DR versus AL mutants using IPA revealed an inverse response compared to Ercc1Δ/−_AL versus WT_AL mice, in line with DR counteracting the accelerated aging effect in Ercc1 mutants (
DR differentially regulates translation dynamics in Ercc1Δ/− as compared to WT
We next examined changes to the expression of translation initiation factors and ribosomal subunits in the context of DR in our ribosome profiling data. DR increased the expression of translation initiation factors in both WT and Ercc1Δ/− mice (
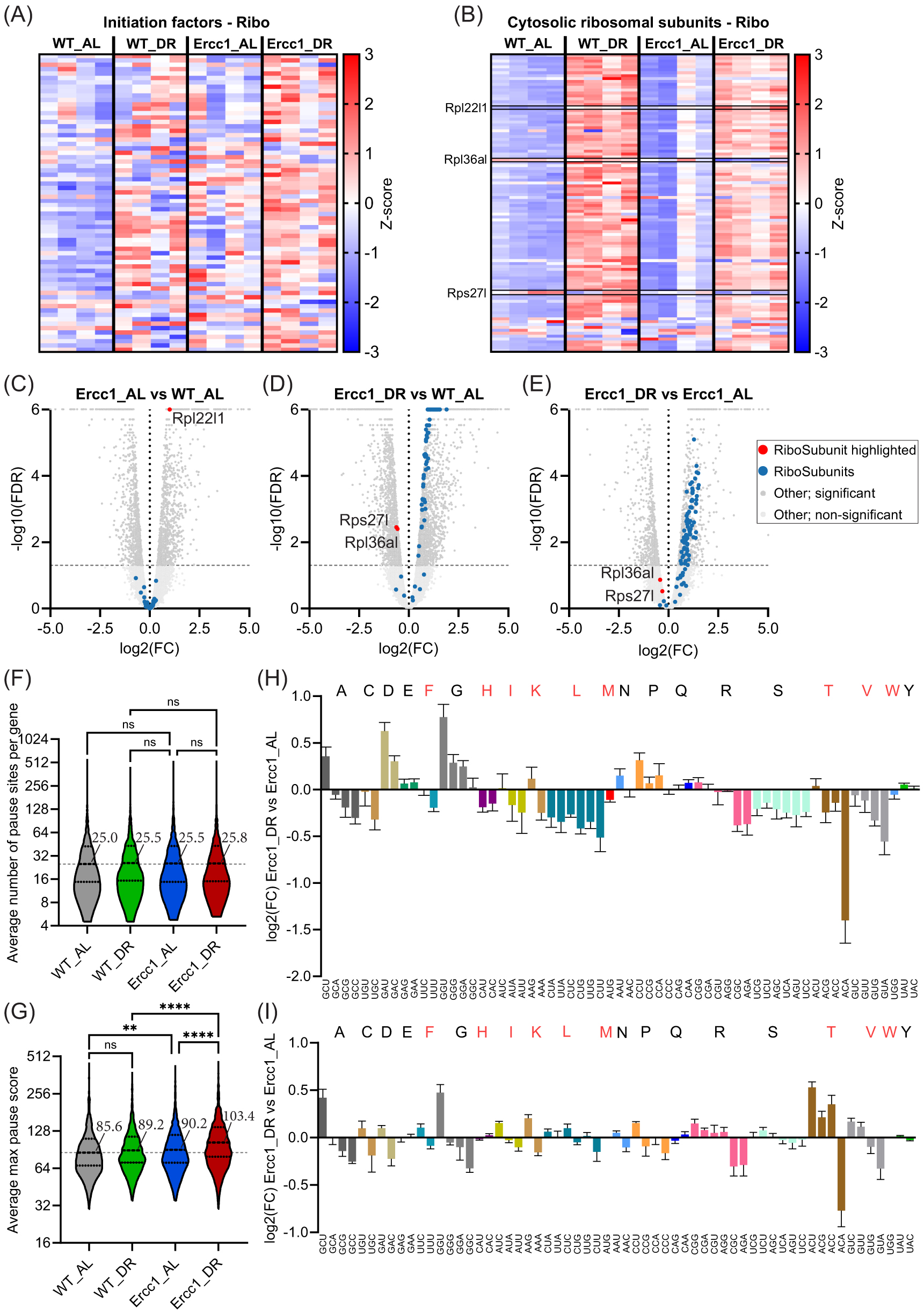
(A,B) Heatmaps of ribosome profiling expression data for initiation factors (A) and cytosolic ribosomal subunits (B), with red indicating an upregulation and blue a downregulation. Relevant factors are highlighted. (C–E) Vulcano plots of cytosolic ribosomal subunit expression in ribosome profiling data for the effect of Ercc1Δ/− against WT (C), the effect of DR in Ercc1Δ/− against AL-fed WT mice (D), and the effect of DR in Ercc1Δ/− against AL-fed Ercc1Δ/− (E). Relevant factors are highlighted. (F,G) Violin plots displaying the mean number (F) and median intensity (G) of ribosomal pause site scores averaged per group. (H,I) Bar graph plot showing log2 fold changes of absolute (H) and aa normalized (I) p-site codon occupancy in DR-fed Ercc1Δ/− mice compared to AL-fed Ercc1Δ/−. Essential amino acids are indicated in red. Error bars depict SD. Significance levels are denoted "ns" for p > 0.05, ** for p ≤ 0.01, and **** for p ≤ 0.0001, with comparisons made to indicated control groups.
Investigating the changes in cytosolic ribosomal subunits, we discerned an atypical expression pattern for Rpl22l1 as the only significantly increased cytosolic ribosomal subunit protein in Ercc1Δ/− mice when compared to WT (
Continuing our examination of translational dynamics, we found that the number of pause sites per gene showed a homogenous distribution for all groups with no statistical differences (
DR further downregulates uORF expression in Ercc1Δ/− mice
In addition to the DR-induced changes in translational dynamics, DR drastically increased the number of differentially expressed uORF-containing transcripts in Ercc1Δ/− mice beyond the changes already triggered in AL-fed conditions (
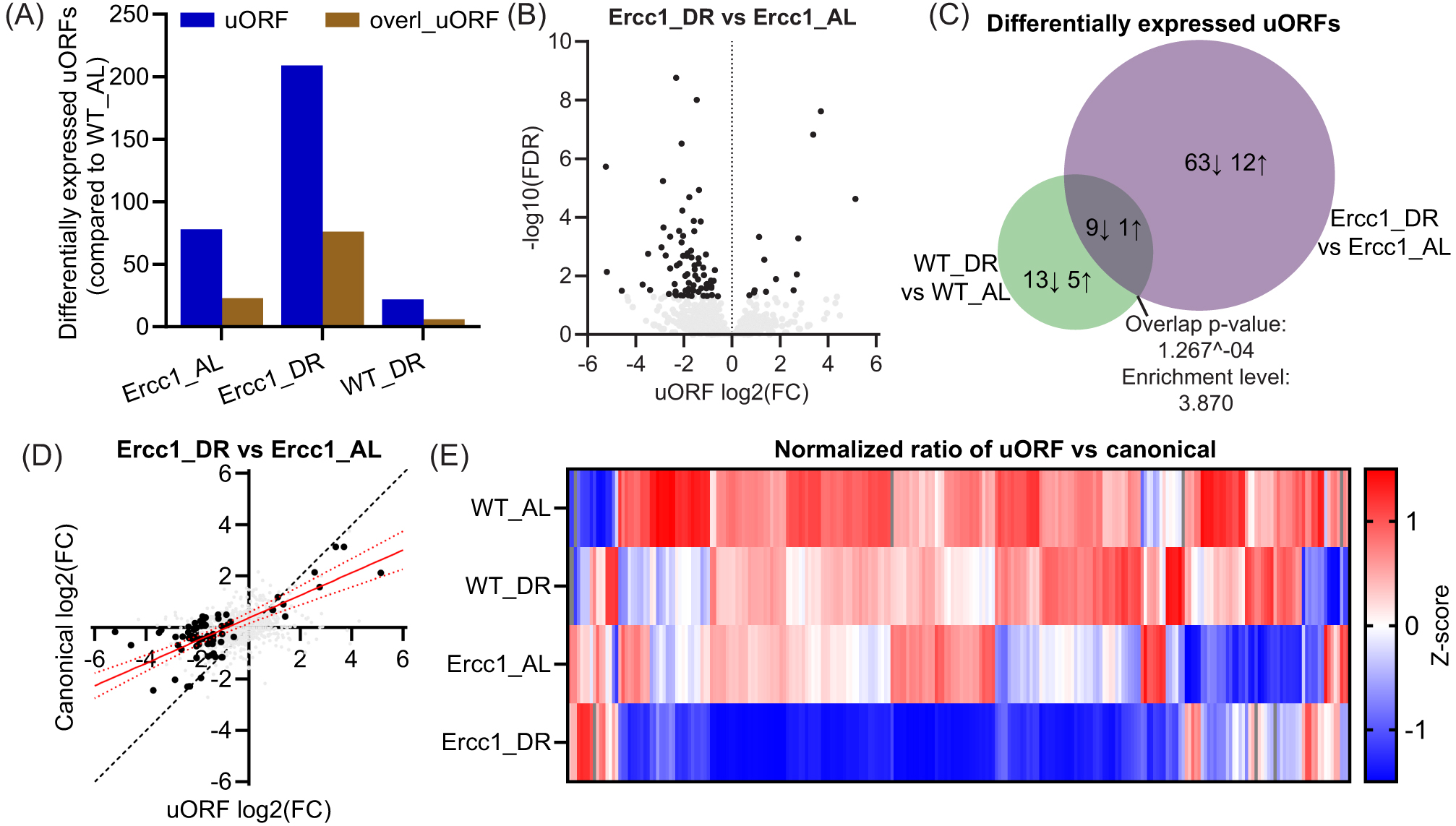
(A) Bar graph displaying the number of differentially expressed uORFs and overlapping uORFs in Ercc1Δ/− AL or DR-fed and WT DR-fed against WT AL-fed mice. (B) Vulcano plot of uORF expression altered by DR in Ercc1Δ/−. Black dots are significant, while gray dots are not. (C) Venn diagram of differentially expressed uORFs due to DR in WT and Ercc1Δ/− genetic backgrounds. Arrows indicate activation or inhibition of uORF sequence translation. (D) Scatterplot illustrating the relation between uORF and canonical fold change differences between DR and AL-fed Ercc1Δ/− mice, with significantly altered uORFs being depicted in black. A regression line with a 95% confidence interval, based on the significant uORFs, is represented in red. (E) Heatmap representing the relative ratio between uORF and canonical ORF expression based on all uORFs that were differentially regulated between WT_AL and at least one other group. Red means upregulated, blue means downregulated, and gray means that no direction could be calculated.
As described earlier, the ratio of uORFs to their canonical ORF expression was reduced in Ercc1Δ/− mice. DR further reduced this ratio in Ercc1Δ/− mice, but only marginally in WT mice (
Discussion
Aging induces a variety of changes, including genome-wide transcriptional decline, preferentially affecting long genes, demonstrated first in progeroid DNA repair-deficient Ercc1Δ/− mice and later also with natural aging in numerous species, strengthening the relevance of accelerated aging for understanding normal aging4–7,54,55. Using the liver as a well-characterized organ central to metabolism, we investigated here whether and to what extent these transcriptional changes are counteracted at the translational level or whether translation degenerates as well. Our findings indicate that transcriptional output is largely retained at the translational level in both Ercc1Δ/− and WT mice, regardless of diet or age. In the liver of Ercc1Δ/− mice we observed significant alterations in processes linked to inflammation and metabolism. Many of these responses are stress-related56,57 and likely triggered by sterile inflammation in response to DNA damage7,58–60. Some consequentially altered metabolic pathways could be detrimental. For example, increased AhR signaling activity has been inversely linked to NAD+ metabolism61,62, while NAD+ supplements have been shown to extend the health and lifespan of mice, including DNA-repair-deficient models63–67. Interestingly, many alterations were influenced in an age-related, DNA-damage-dose-dependent manner, while 30% DR ameliorated a variety of tissue-damage-related pathways in Ercc1Δ/− mice, findings that are logically consistent with the anti-aging benefits exerted by DR5. Notably, DR influenced translational regulators “eIF2 signaling” and “regulation of elF4 and p70S6K signaling”50,68,69, which were predominantly enriched for ribosomal subunits. The increased phosphorylation of eukaryotic translation initiation factor elF4 has recently been linked with translation of mRNAs involved in fasting-induced lipid catabolism and ketone body production via a newly discovered lipid-induced AMPK–MNK–eIF4E kinase signaling pathway50, which aligns with the observed differential regulators and pathway expression in our study.
The distinct expression of certain cytosolic ribosomal subunits under conditions of DNA damage and DR draws attention to potentially unique functions related to translation. Increased expression of Rpl22l1 during WT and accelerated aging is noteworthy, as it has previously been implicated in processes like proliferation, translation, and epithelial-to-mesenchymal transition70–72, likely linked to damage-induced cellular turnover. Moreover, DR modified Rps27l expression, which has previously been associated with p53 signaling and autophagy53,73,74. Concurrently, Rpl36al has been postulated to play a role in peptide bond formation and is the only ribosomal protein so far identified that can engage with tRNA, regardless of its ribosomal association75. Notably, DR specifically influenced the expression of Rps27l and Rpl36al during conditions of DNA-damage-driven cellular stress but not in young WT, suggesting a synergistic requirement of DNA damage accumulation and nutrient stress.
Curiously, the translation of ribosomal subunits in the liver of naturally aged C57BL/6 mice (32 months) has previously been reported to decrease through inhibition of 5’-TOP-related genes16. In contrast, our results demonstrate that DR can increase both the transcription and translation of the translational machinery, while other studies in different animals have reported similar increases at the transcriptional level10,76. The reason for this discrepancy is yet unknown but might be related to the improved health in mice with a more outbred-like C57BL6J/FVB hybrid background as compared to the pure C57BL6J strain. Future work might compare relevant datasets to exclude other aspects such as differences in data handling, normalization, or validate findings more at an absolute quantitative level.
The general upregulation of initiation and cytosolic ribosomal subunits could reflect an attempt to increase ribosomal turnover, enhancing translational regulatory oversight and fidelity, or might be an apparent increase due to the relative nature of sequencing when total transcription and translation decreases. In addition, the absence of changes in mitochondrial ribosomal subunits presents a striking dichotomy, suggesting a disconnect between the regulatory adjustments of cytoplasmic and mitochondrial ribosomal subunits under DR, warranting further examination, also considering the reduced correlation in age-related changes in transcriptome versus proteome10.
Furthermore, translation dynamics were also influenced, with pause site intensity, absolute codon occupancy, and uORF expression being more strongly affected by ERCC1-deficiency than by WT aging, consistent with known phenotypical differences27,63. Moreover, DR in young WT mice showed little effect on various aspects of translational dynamics, while having a strong impact on Ercc1Δ/− mice, further supporting the idea of synergistic interactions in responses between genotoxic- and nutrient-induced cellular stress. Codon occupancy showed a clear discrepancy between the effects caused by the Ercc1Δ/− mutation and DR, with DR reversing the pattern toward AL-fed WT levels. Although correlations with essential amino acid and codon usage were observed, direct mechanistic explanations remain unidentified.
Both relative codon occupancy and ribosomal pausing are linked to ribosomal movement, yet they displayed discordant behavior. However, pause score intensity and relative uORF changes were both concordantly intensified by DR in Ercc1Δ/− mice, suggesting that these processes are potentially beneficial adaptive mechanisms to ensure efficient protein synthesis despite DNA damage and nutrient scarcity. Ribosomal pausing is, for example, an important aspect of translation, affecting protein folding, localization, fidelity, and protein expression by altering the speed of translation elongation19,77. However, exacerbation of pausing, that is, ribosomal stalling, during aging has been suggested to disrupt co-translational proteostasis and induce detrimental age-related translational reprogramming, potentially leading to reduced levels of vital proteins and an exacerbation of the aging phenotype in neurological tissues, complicating its mechanistic role17,22,78. It remains to be investigated if DNA damage could be an antagonistic pleiotropic regulator, where mild levels of genotoxic stress reduce the speed of translation, while excessive damage drives proteotoxic stress, and whether these effects could be tissue dependent3,24,79.
A large number of canonical ORFs associated with the uORFs we observed are known to produce proteins crucial for cellular homeostasis. The relative reduction of these uORFs, compared to their canonical counterparts, suggests a protective or adaptive mechanism. Interestingly, uORF-related gene transcripts can also contain other regulatory elements like internal ribosome entry sites (IRES), which are often found in mRNAs coding for master regulators of cellular homeostatic responses. These mRNAs must be tightly controlled, and under various stresses, including DNA damage and amino acid starvation, the relative increased selective translation of these sequences ensures homeostasis80. Moreover, while the observed uORF changes are statistically significant, the relatively low number of uORF-related footprints and their potential interaction with IRES elements suggests that further research, potentially involving deeper sequencing, may provide additional insights.
Models of accelerated aging often do not fully encapsulate all aspects of natural physiological aging3,24,79,81. However, the Ercc1Δ/− mouse model shows strong similarities with natural aging at both the molecular and physiological level25,82,83 and similarly displays accelerated glycan- and epigenetic aging clocks across multiple tissues84–86. In the current study, differences between accelerated and naturally aged mice were mainly characterized by lipid metabolism. Given the central role of lipid metabolism in longevity, these differences may be partially explained by survival-bias-driven metabolic adaptations in the naturally aged mice, distinguishing them from the broader cohort.
In previous work, we have shown that aging in both Ercc1Δ/− and wild-type mice is correlated with a gene-length-dependent transcriptional decline due to physical stalling of elongating RNA polymerases leading to a disbalance between long and short genes4. In the present study, we found a high similarity between transcriptional and translational profiles, suggesting that most of the gene-length-dependent transcriptional changes are likely “translated” to the protein level. Another feature, which is widely associated with aging but has remained largely elusive, is a progressive decoupling between the transcriptome and proteome10,87,88. At first sight this might seem difficult to rationalize with our observations; however, continuation of a gene-length-dependent disbalance onto the protein level might lead to incomplete multi-subunit protein complexes when one subunit derived from a larger gene is under-expressed due to the transcription mentioned above. Incomplete protein complexes can in turn trigger the unfolded protein response and induce degradation of the affected subunits89,90 or trigger compensatory mechanisms91. Since cells contain numerous protein complexes, this might partially explain the disconnect between the transcriptome and proteome during aging. Future studies should investigate whether gene length–dependent transcriptional decline contributes to the loss of proteostasis during aging to evaluate the validity of this hypothesis. Moreover, the role of RNA damage-driven ribosome collision herein and its contribution to aging and related changes remains to be further investigated92–94.
In conclusion, our data showed that DR in Ercc1Δ/− mice elicited a multifaceted adaptive response, revealing alterations across several molecular pathways, including inflammation, protein synthesis, and cellular metabolism, while demonstrating the adaptive capacity of the translational machinery under conditions of stress and nutrient limitations. Moving forward, the influence of dietary composition, RNA homeostasis, and the proteome should be examined to shed further light on the interplay between genomic instability, metabolism, translational control, and lifespan.
Methods
Mouse models and ethics statement
Male Ercc1Δ/− and wild-type (WT) littermate control (Ercc1+/+ ) mice were obtained by crossing Ercc1Δ/+ (in a pure C57BL6J or FVB background) with Ercc1+/− mice (in a pure FVB or C57BL6J background, respectively) to yield Ercc1Δ/− offspring with a genetically uniform F1 C57BL6J/FVB hybrid background. These two backgrounds were crossed to minimize unfavorable characteristics, like early onset of blindness in an FVB background or deafness in a C57BL6J background. Since Ercc1Δ/− mice were smaller, food was administered within the cages, and water bottles with long nozzles were used from around two weeks of age. Experiments were performed in accordance with the Principles of Laboratory Animal Care and with the guidelines approved by the Dutch Ethical Committee in full accordance with European legislation (DEC no. 139-12-13 and 139-12-18).
Housing conditions and dietary regimens
Animals were housed individually in ventilated cages under specific pathogen-free conditions, in carefully controlled environments (20–22 °C, 12h light:12h dark cycle). Mice were visually inspected daily and weighed weekly and scored blindly for gross morphological and motor abnormalities. All animals were bred and maintained on AIN93G synthetic pellets (Research Diet Services B.V., Wijk bij Duurstede, The Netherlands; gross energy content 4.9 kcal/g dry mass, digestible energy 3.97 kcal/g). On average, 6–16-week-old Ercc1Δ/− mice ate 2.3 g of food per day. The animals that were given dietary restriction (DR) were so from 7 weeks of age, when development was largely completed, starting with 10% restriction. DR was gradually increased weekly by 10% until 30% DR (1.6 g/day), which was provided from 9 weeks of age onward as previously published5. Adult WT mice ate on average 3.0 g of food per day, resulting in 2.1 g/day for 30% DR and a reduction in body weight (Fig. S1F). To avoid alterations in the biological clock, food was provided to the animals just before the start of the dark (active) period, Zeitgeber Time (ZT) 12:00, except for the day of sacrifice, when no food was given prior to collect.
At 16 weeks of age, WT and Ercc1Δ/− mice on AL and DR regimens were sacrificed at the beginning of the dark period between ZT13 and ZT16. In addition, a group of AL-fed 7-week-old Ercc1Δ/− mice was included, representing a biologically younger age and as a control for the start of DR, and a group of AL-fed 104-week-old WT, for comparison to natural aging (Fig. S1A). Prior to weaning, animals were randomly divided over all groups to prevent selection bias. Animals were euthanized when necropsy age was reached, livers were snap frozen in liquid nitrogen, and stored at −80°C for molecular analysis.
mRNA sequencing
Snap-frozen liver specimens were lysed in QIAzol lysis reagent with the TissueLyser LT (QIAGEN), and RNA was isolated using the miRNeasy mini kit (QIAGEN; 217004) according to the manufacturer’s protocol with optional on-column DNase I treatment (QIAGEN; 79254). RNA quality and quantity were assessed using the NanoDrop One (Thermo Fisher Scientific, USA) and Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA; G2939BA), respectively. RNA integrity values ranged between 8.4 and 9.5. Samples were sent for mRNA paired-end 2*150 bp sequencing (Novogene, Cambridge, United Kingdom) on the Illumina NovaSeq6000 platform to a depth varying between 39887891 and 63618955 total reads per sample.
Ribosome profiling
Snap-frozen tissue samples from the same livers as used for mRNA sequencing were used to perform ribosome profiling with minimal modifications as previously described95,96. Samples were powdered with a BioSpec Pulverizer (#59012MS) on dry ice. Pulverized material was lysed with 500 μl ice-cold lysis buffer (20 mM Tris-Cl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 0.1% IGEPAL CA-630 (Sigma-Aldrich, USA), 1 mM DTT, 10 U/ml RNase-free DNase 1 (Lucigen, USA), 100 μg/ml cycloheximide (Sigma-Aldrich, USA)) for 15 minutes on ice, triturated ten times through a 23G needle, and clarified through centrifugation at 20000 g at 4 °C for 10 min. Total RNA content of sample lysates was estimated using the Qubit™ RNA broad range (BR) Assay Kit (Thermo Fisher Scientific, USA) on an Invitrogen™ Qubit™ 4 fluorometer (Thermo Fisher Scientific, USA). Lysates were then digested in 200 μl aliquots with RNAse I (10 units per 20 μg of RNA; Lucigen, USA) for generation of ribosome-protected fragments (RPFs) at 20 °C for 45 min while shaking at 400 RPM on a thermomixer. The digestion reaction was stopped by adding 5 μl (5U) of SUPERase*In RNAse inhibitor (Thermo Fisher Scientific, USA) and placing the samples on ice. Digested lysates were transferred to Microspin S-400 HR sephacryl columns (Sigma-Aldrich, USA) equilibrated with 3 ml of cold RNase-free polysome buffer (20 mM Tris-Cl pH 7.4, 150 mM NaCl, 5 mM MgCl2) and centrifuged at 600 g at room temperature for 2 min. 10 μl of 10% SDS was added to the digested lysates, and RPFs were extracted using 3 volumes (660 μl) of Trizol LS (Fisher Scientific, USA), followed by the addition of 1 volume (880 μl) of ethanol and RNA purification using the Zymo Direct-zol RNA micro prep kit (Zymo Research, USA) with slight modifications to the manufacturer’s instructions: Columns were spun dry for 1 min at 12000 g, and isolated RPFs were eluted in 20 μl nuclease-free water (Sigma-Aldrich, USA).
Depletion of rRNA was performed using RiboPOOL technology (siTOOLs Biotech; riboPOOL ribo-seq cat# dp-K012-000052, Germany) with a slight modification to the manufacturer’s instructions: 200 pmol of RiboPOOL and 100 μl of beads were used per sample. Next, RNA was purified using the Zymo RNA Clean and Concentrator-5 kit (Zymo Research, USA) and size-selected through denaturing PAGE using 15% TBE-Urea gels (Thermo Fisher Scientific, USA). RNA fragments corresponding to 26–34 nucleotides were excised and recovered from gel slices by crushing the gel and rocking at 20 °C, 700 RPM for 2 hours on a thermomixer. RNA solutions were transferred to Costar Spin-X filter tubes (Thermo Fisher Scientific, USA) and filtered through centrifugation at 2350 g for 6 min. 2 μl of GlycoBlue (Thermo Fisher Scientific, USA) and 700 μl of isopropanol were added per aliquot, and RNA was left to precipitate overnight at −80 °C.
Following precipitation, RNA fragments were pelleted by centrifugation at 21130 g at 4 °C for 45 min. Pellets were washed with 1 ml of 80% ice-cold ethanol and centrifuged at 21130 g at 4 °C for 10 min. RNA pellets were air-dried for 3–4 min and dissolved in 60.75 μl nuclease-free water on ice. RNA fragments were dephosphorylated using 30 U of T4 PNK (Lucigen, USA) for 1 h at 37 °C and purified using the Zymo RNA Clean and Concentrator-5 kit (Zymo Research, USA), where isolated RNA fragments were eluted in 9.5 μl nuclease-free water. Purified fragments were ligated to a pre-adenylated 3’ oligonucleotide linker using 100 U of T4 RNA ligase 2 Deletion Mutant (Lucigen, USA) and 5 U T4 RNA ligase 1 (Thermo Fisher Scientific, USA) at 23 °C for 3 h. Leftover linker was removed using 5’ Deadenylase (New England Biolabs, USA) and Rec J Exonuclease (Lucigen, USA). Linker-ligated RNA fragments were reverse-transcribed into cDNA using EpiScript reverse transcriptase (Lucigen, USA). cDNA was treated with Exonuclease I (Lucigen, USA) for 30 min at 37 °C, followed by 15 min at 80 °C with a reduction to 4 °C, and further treated with 5 U of RNAse I (Lucigen, USA) and 2.5 U of Hybridase Thermostable RNase H (Lucigen, USA) at 55 °C for 5 min, followed by an incubation step at 4 °C to stop the reaction. Treated cDNA was purified using the Zymo Oligo Clean and Concentrator Kit (Zymo Research, USA) with modifications to the manufacturer’s instructions: Columns were spun dry for 1 min at 21130 g, and isolated RPFs were eluted in 9.5 μl nuclease-free water (Sigma-Aldrich, USA).
Size-selection of cDNA containing ligated linkers was performed through denaturing PAGE using 10% TBE-Urea gels (Thermo Fisher Scientific, USA). cDNA fragments corresponding to 70–80 nucleotides were excised and extracted with ammonium acetate and SDS, followed by overnight precipitation with isopropanol as described above. Size-selected cDNA was circularized for 3 h at 60 °C using 100 U of circLigase I (Lucigen, USA), followed by heat inactivation for 10 min at 80 °C, and amplified using Phusion high-fidelity polymerase (New England Biolabs, USA) with reverse primers containing unique barcode sequences for 10 cycles of 30 sec at 98 °C, 15 sec at 94 °C, 5 sec at 55 °C, and 10 sec at 65 °C. Following amplification, 5 μl of 3 M NaCl (Thermo Fisher Scientific, USA), 1 ml of ethanol, and 2 μl of GlycoBlue (Thermo Fisher Scientific, USA) were added to each aliquot of cDNA and left to precipitate overnight at −80 °C as described above.
Amplified cDNA libraries were size-selected using 8% non-denaturing TBE-Urea gels (Thermo Fisher Scientific, USA). cDNA libraries corresponding to 150 nucleotides were excised and recovered from gel slices by crushing and subsequent rocking at 37 °C at 700 RPM on a thermomixer for 2 hours. cDNA solutions were transferred to Costar Spin-X filter tubes (Thermo Fisher Scientific, USA) and filtered through centrifugation at 2350 g for 6 min. cDNA libraries were purified using the Zymo DNA Clean and Concentrator-5 kit (Zymo Research, USA) with modifications to the manufacturer’s instructions: Columns were spun dry for 2 min at 12000 g and isolated RPFs were eluted in 13 μl nuclease-free water (Sigma-Aldrich, USA).
cDNA libraries were quantified using Qubit™ DNA high sensitivity (HS) Assay Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions on an Invitrogen™ Qubit™ 4 fluorometer (Thermo Fisher Scientific, USA) and Bioanalyzer 2100 (Agilent) using the High Sensitivity DNA kit and pooled in equimolar ratios. Sequencing was performed on a NextSeq2000 (Illumina; 1×50 bp) at the Utrecht Sequencing Facility (USEQ) to a depth varying between 37831148 and 70879824 total reads per sample.
mRNA- and Ribo-seq data processing
Quality control of Ribo-seq data was performed by filtering reads for mitochondrial, rRNA, and tRNA sequence contaminations using Bowtie2 v2.4.2 (default settings, except; --seedlen 25)97. The resulting ribosome-protected fragments were consecutively aligned to the genome with STAR (version 2.7.3, default settings, except; sjdbOverhang 29, --outFilterMismatchNmax 2, --outSAMattributes = All, --outSAMtype SortedByCoordinate, --limitOutSJcollapsed 10000000, --limitIObufferSize 300000000, --alignSJoverhangMin 1000). RiboseQC v1.198 (default settings, except; read_subset = False, fast_mode = False) was used to check the quality of Ribo-seq samples and to extract footprint length distributions, periodicity (3 nt codon movement), and P-site counts from uniquely mapped reads. Quality control of mRNA data was performed with with FastQC v0.11.9.
For subsequent expression analysis, mRNA reads were treated as single-end and truncated to 28 nt to imitate ribosome-protected fragments. Next, both mRNA and ribo-seq datasets were treated identically, and processing of raw sequencing reads was performed on our in-house-generated data analysis pipeline. Removal of sequence adaptors, tRNA, and rRNA from both mRNA- and Ribo-seq and truncation of mRNA reads was performed using Trimmomatic (version 0.39). Trimmed reads were aligned to the coding region (CDS) of the mouse reference genome (annotation: gencode.vM20.annotation.gtf; genome: GRCm38.p6.genome.fa; http://gencodegenes.org/mouse/release_M20.html) using STAR (version 2.7.3). Read counts for each gene were obtained using FeatureCounts (as part of SubRead version 1.6.4), filtered using filterByExpr with count set to 10, followed by TMM normalization, quantification of log fold changes (logFC), and false discovery rates (FDR) using EdgeR (version 3.32.1). Statistically significant changes in mRNA- and ribo-seq data are referred to as differentially expressed genes (DEGs), depending on context.
For ORF quantification, ribosome-profiling adapters were clipped from reads and filtered for the standard quality threshold used by CutAdapt v3.499. Fragments shorter than 25 nucleotides were discarded, and reads were filtered for contaminants using bowtie v2.4.2 as described above. The RPFs were mapped to the mouse genome (annotation: gencode.vM20.annotation.gtf; genome: GRCm38.p6.genome.fa; http://gencodegenes.org/mouse/release_M20.html) using STAR v2.7.8a and analyzed with ORFquant using default settings100. All CDS regions from the ORFquant output were analyzed for potential P-sites across all three frames by a custom Python script. In-frame P-sites were retained, yielding one nucleotide (NT) position per codon. An intersection operation was performed on the resulting BED file with the actual P-sites per sample calculated by ORFquant, providing the number of P-sites present per transcript per sample. Finally, logFC and FDR were calculated with EdgeR (version 3.32.1). Example images of canonical ORF and uORF translation were generated using the Integrative Genomics Viewer (IGV) by normalizing coverage data calculated by ORFquant101.
Data analysis
Principal Component Analysis (PCA) was employed on the data normalized and mapped to coding sequences (CDS). We used the R Bioconductor packages “prcomp” and “gplots” for PCA analysis and plot generation, respectively. Enrichment analysis of pathways and upstream regulators was conducted using overrepresentation analysis (ORA). This process was facilitated through the Interactive Pathway Analysis (IPA) software (Ingenuity Systems, QIAGEN), designed to manage complex genomics data. For these analyses, we utilized a pre-filtered list of differentially expressed genes, selecting those with an FDR ≤ 0.05 and log(FC) > |0.5| to denote differential expression. Upstream regulator results were filtered to only include genes, RNAs, and proteins while excluding, for example, drug compounds, with Z-scores indicating the likely activation state of biological function. The identification of over-represented canonical pathways was achieved based on the data in the Ingenuity Pathways Knowledge Base. In IPA, gray bars represent pathways where activity predictions are not possible, while white bars indicate pathways with z-scores near zero or those ineligible for analysis due to fewer than four analysis-ready molecules (z-score = NaN).
GSEA (v4.3.2—build 13, standalone version) was run using TMM-normalized CPM values as input. For each comparison between groups (as shown in the figures), genes were ranked by signal-to-noise ratio, and enrichment scores were then calculated for predefined gene sets comprising annotated mitochondrial and cytosolic ribosomal subunits and translation initiation factors (Supplemental file 1)102. Collapse/Remap to gene symbols was set to “No_collapse,” and permutation type was set to “gene_set” to improve workflow for NGS data. All other settings were left at default.
Differential translational efficiency was examined using the DeltaTE software tool with standard settings and “batch” set to 0103. Genes were deemed differentially translated if they exhibited an FDR≤0.05. Pause site prediction and P-site codon occupancy were analyzed by the respective modules from Ribotoolkit using default settings35. Pause site prediction was powered by PausePred, which calculates the number of reads mapped to each position within a sliding window of length 1000 (step = length/2), considering footprints of 28–30 nt, normalizes these values over the average read density within the window, and computes the pause score as the average across overlapping windows104. For quantification of pause scores, those genes were used for which a pause score could be calculated in all genes using a default pause score threshold of 20 (n = 1228 genes). P-site codon occupancy was calculated with ribotoolkits and referenced as “absolute codon occupancy,” as values are not automatically normalized for amino acid abundance. After normalization for differences in amino acid usage outside of ribotoolkits, the data was referenced as “relative codon occupancy.” Heatmaps were based on edgeR normalized data and z-score transformed for visualization. Heatmaps based on ratio comparisons were log2 transformed before z-score transformation.
Statistics
Statistical analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA, USA; version 9.2.0), DeltaTE103, Ribotoolkit35, IPA (Ingenuity Systems, QIAGEN), GSEA (v4.2.3 Build 10), or R Studio (2021.09.1, build 371). Significant p values were expressed as *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. All data with error bars are presented as mean ± standard deviation (SD) as specified in figure legends. Nonparametric statistical analysis was applied to pause score analysis, as the data was not normally distributed. Statistical differences in pause score violin plots with two groups were calculated using the Mann–Whitney unpaired test in GraphPad Prism. Statistical differences in pause score violin plots comparing >2 groups were calculated using one-way ANOVA, and multiple comparisons testing was corrected for using Dunn’s test, also in GraphPad Prism. Statistical significance and enrichment levels for the overlapping sections of the Venn diagrams were calculated using a custom R script based on hypergeometric distribution, as previously described105. Pie chart statistics were calculated based on cumulative binomial distribution probabilities.
Acknowledgments
We are grateful to Renata Brandt, Sander Barnhoorn, and the animal caretakers for general assistance with mouse experiments and Irene van Dijken and Willianne Vonk for experimental support. This research was funded by ONCODE, supported by the Dutch Cancer Society. J.H.J.H. was additionally supported by the National Institute of Health (NIH)/National Institute of Aging (NIA) (AG17242), the European Research Council Advanced Grant Dam2Age, and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation—Project-ID 73111208—SFB 829); J.H.J.H. and W.P.V. by Memorabel (ZonMW 733050810), BBoL (NWO-ENW 737.016.015), and the European Joint Programme Rare Diseases (TC-NER RD20-113); and W.P.V. by the Regiodeal Foodvalley (162135) and the ADPS Longevity Research Award.
Author Contributions
IvG, JHJH, and WPV conceptualized and designed the study. IvG, SAGE, and KS performed wet lab experiments. IvG, RAO, DAF, and JvD performed dry lab experiments and statistical analysis of data. IvG, SvH, JHJH, and WPV wrote this article, and RAO, DAF, JvD, SAGE, and KS contributed to editing this article. All authors contributed to the article and approved the submitted version.
Data Submission Statement
Raw data files for ribosome profiling and mRNA-seq have been submitted to the NCBI Gene Expression Omnibus (GEO) under accession numbers GSE288421 and GSE288427, respectively.
Conflicts of Interest
The authors declare that they have no actual or apparent conflict of interest between authorship of this study and any other activities.
AI Statement
Generative AI tools (ChatGPT4o/Claude 3.5 Sonnet) were used for minor improvements to grammar and sentence structure, but not for data acquisition or analysis. The authors reviewed and edited AI-generated changes and take full responsibility for the content of this publication.
Supplementary Materials
Supplemental information can be found here: Supplementary.